|
No. 1
Mechanism and Modeling of Human Disease-Associated Intronic Variants that Perturb RNA Splicing
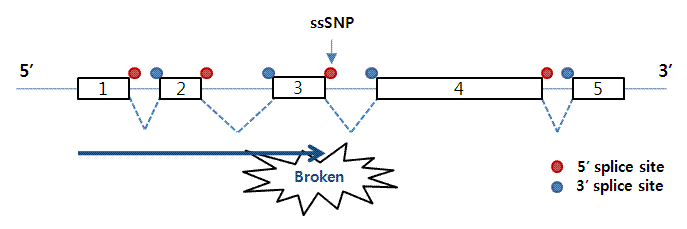
It is estimated that 10-30% of disease-associated genetic variants affect splicing. Splicing variants may generate deleteriously altered gene product and are potential therapeutic targets. However, experimental diagnosis for splicing variants is time-consuming and reliable computational prediction tools have not been established, especially for the near-exon intronic splice region. The major challenge lies in the redundant and ill-defined branch sites and other splicing motifs therein. Here, we carried out unbiased massively parallel splicing assays on 5,307 disease-associated variants overlapped with branch sites and collected 5,884 variants across the 5’ splice region. From the 11% variants significantly altering 3’ss splicing, we identified two distinct splicing altering mechanisms, novel splice site competition and branch site dysfunction, whereas the splicing altering mechanism of the 5’ intronic variants is mainly the splice site destruction. Statistical learning combined with these molecular features allows precise prediction for altered splicing from an intronic end variant. This statistical model provides identity and ranking of biological features that determine splicing, which serves as transferable knowledge for future studies, and out-performs the benchmarking predictive tool. Moreover, we demonstrated that intronic splicing variants may associate with disease risks in human population. Our study elucidates the mechanism of splicing response of intronic variants, which classify disease-associated splicing variants for the promise of precision medicine.
在後基因體時代,次世代基因定序蓬勃發展,然而對基因變異的解讀卻遠遠落後。10-30%的致病突變是核醣核酸的剪接突變。為了基因檢測與精準醫療的發展,我們必須發展解讀和預測剪接突變的工具。在內含子之中,又以近剪接點的區域為剪接位點本身外最重要的剪接調控區域,但是內含子中的剪接元件通常有多個且定義模糊,無法簡單預測其突變的結果。這也是為何目前主流的剪接突變預測工具對內含子的突變幾乎莫可奈何的原因。為了系統化解釋突變對剪接的影響,也探討剪接對疾病發生的機制,我們著手開發系統化偵測剪接突變的高通量實驗,同時測量在人類疾病資料庫中登記的上萬個內含子突變。我們將從實驗結果擷取影響剪接決定的因子,例如演化保守度和與其他調節分子結合位點的改變,以統計模型解讀影響剪接決定的因素,此模型可用於預測其他內含子突變對剪接的影響,判斷是否新生剪接位點,以及最終的剪接效率。此外,我們還將在台灣人體生物資料庫中預測內含子剪接突變,用生化實驗與遺傳流行病學探討這些突變與疾病的關聯。總和來說,我們將結合跨領域的專長,包含分子生物學、基因體學與統計資訊學,來發展可偵測內含子剪接突變的演算法與網路工具,為精準醫學正確註釋剪接突變。 |
No. 2
Study translational control of diseases related SNPs in 3’ UTR
Genome-wide association study has focused on the change of amino acid in the coding region and/or the transcriptional regulation of gene expression. The translational control has been overlooked partly due to the absence of the protein expression profile. However, post-transcriptional regulation determines the level, the location and the timing of protein expression. Therefore, I propose to study how SNPs affect gene expression translationally with a massive parallel assay. From the publicly available database, such as dbSNP, Exome Aggregation Consortium (ExAC) data and The Cancer Genome Atlas (TCGA), we can identify SNPs and mutations overlapping with the microRNA and the RNA-binding protein binding sites in the 3’ UTR. We will then clone 3’ UTR of the minor allele and their corresponding major allele into a translation reporter, such as a luciferase reporter, and test how these minor alleles affect the protein expression. More specifically, 3’UTRs with SNPs/mutations are cloned after luciferase coding region and within sequencing adaptors. These sequences are then in vitro transcribed, transfected into cell lines (HEK293, HeLa and SH-SY5Y neuronal cell lines). The transcripts level before and after transfection is compared to calculate the transcript stability. The luciferase reading normalized to the transcript level at harvest is compared for translation efficiency. Further analysis will be carried out to dissect the effect of SNPs/mutations that interrupt microRNA binding and RNA-protein interaction. I expect to learn how SNPs/mutations at 3’ UTR regulate RNA stability and translation efficiency. This study will greatly help us to understand the protein expression regulation in physiological and pathological background.
|

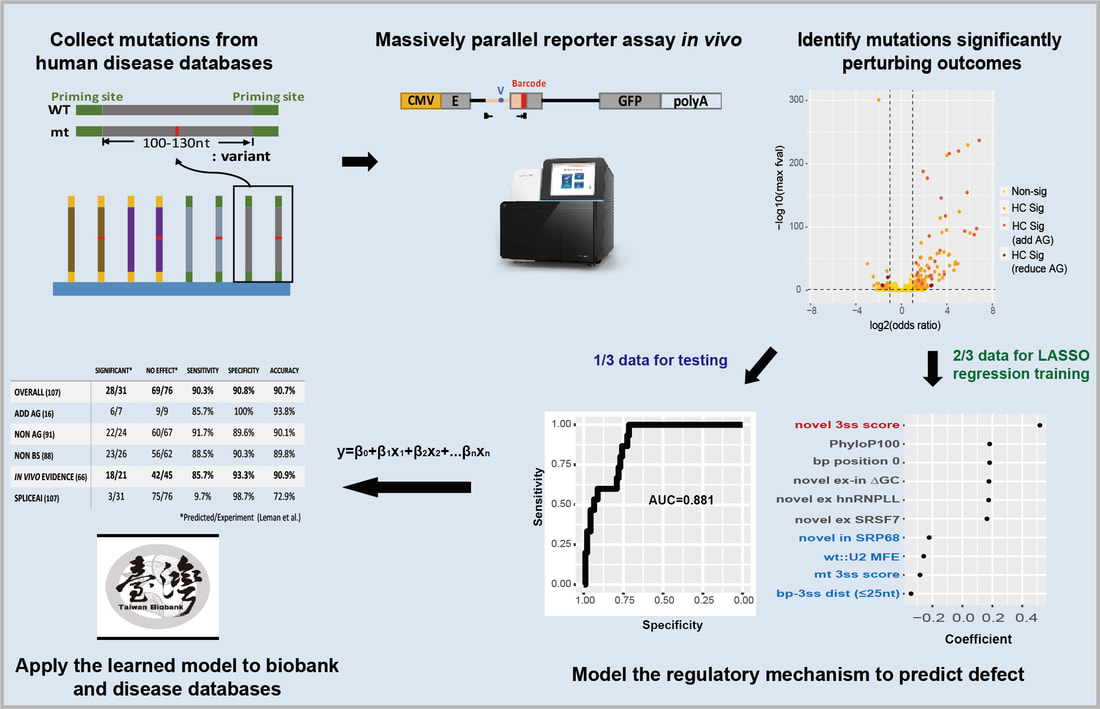

Interdisciplinary approaches
We seek to use computational and biochemical approaches to study post-transcriptional gene regulation, including pre-mRNA splicing, mRNA stability, translation efficiency and its degradation.
|
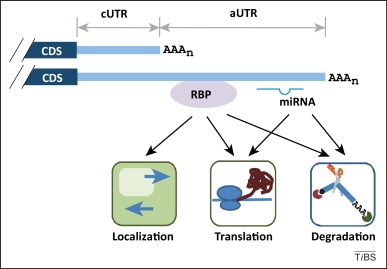
UTRs (untranslated regions) are involved in gene regulation.
Trends in Biochemical Sciences 2013 38, 312
|
